Whole (16 page)
Authors: T. Colin Campbell

My team’s task was to learn something about the climatic and geographic conditions that fostered
Aspergillus flavus
growth. We studied several edible plants, but focused specifically on peanuts.
Shortly thereafter, the dean who hired me at Virginia Tech, Charlie Engel, asked me to join him in developing the nationwide childhood nutrition program in the Philippines in collaboration with Manila’s Department of Health—a project funded by USAID. One of our main goals was to identify a source of protein for these children that could be grown locally and relatively inexpensively. The obvious answer, at least to us, would have been peanuts. They’re high in protein, most kids love them, and they grow like crazy in a wide variety of climates and settings. There was just one problem: AF.
Before we could grow peanuts to solve the protein gap, we had to understand and solve the potential AF contamination problem. Because of my earlier experience with AF, that became my assignment. After setting up and equipping an analytical laboratory in Manila, I then began with my colleagues in the Philippines to explore the chief food sources of AF consumption. Were peanuts the main source of contamination? What about other foods? Did the people eating AF-contaminated foods really get more liver cancer? If so, what could we do to eliminate AF, or
neutralize its negative effects, so we could use peanuts as a cost-effective protein source for the poor?
We started by collecting peanut products from the marketplace. Shelled peanuts, the more expensive product purchased by the affluent (our original samples came from a cocktail party at the U.S. Embassy!) were clean, with little or no AF. In contrast, peanut butter, a cheaper product especially consumed in urban centers like Manila, was heavily contaminated. All of the twenty-nine peanut butter samples we initially collected contained AF, with an average of 500 parts per billion (ppb),
6
but with exceptional levels as high as 8,600 ppb.
7
These findings were alarming because, at that time, the U.S. FDA had proposed an upper limit of 30 ppb as a “safe” level in human food (later revised downward because even lower levels were shown to cause serious toxicity and cancer in rats, rainbow trout, and very young ducklings).
8
To learn the reasons for this huge discrepancy in AF levels between whole cocktail peanuts and peanut butter, I joined the Philippines’ FDA Commissioner in a visit to a peanut butter manufacturing plant. The answer was easy to see. In the manufacturing plant, whole peanuts in their shells were placed onto one end of a conveyer belt, which moved past a line of workers; at the end of the moving belt, the peanuts were delivered into a grinder and a big cooking pot. As the peanuts passed by the workers, they handpicked kernels for the cocktail peanuts, leaving the rest to be dumped into the grinder and cook pot to make peanut butter. The good, attractive kernels went into the cocktail jars, the bad into the peanut butter tank. By “bad,” I mean the discolored, often shriveled kernels—the ones most likely to be infected with the fungus. These kernels, we learned when we tested them, contained AF in concentrations as high as two million parts per billion, meaning that even a single fungus-contaminated kernel could spoil an entire batch of peanut butter and easily push AF levels over the allowable limit.
9
With additional funding from the National Health Institute, I then did a quick survey of possible consumers of AF and learned that, just like in the United States, children ate most of the peanut butter in the Philippines. Because I assumed that virtually all commercially sold peanut butter was contaminated, my coworkers and I then visited homes to ask whether they customarily ate peanut butter and, if so, whether we could purchase any partly emptied jars for AF analysis. We also asked the mother in the
household for an estimate of when and how much peanut butter had been consumed in the previous twenty-four to forty-eight hours, and from this I estimated actual AF consumption. We also collected urine specimens from each family member so that, for future follow-up studies, we might be able to measure some product of AF in the urine as a reliable marker of AF ingestion.
10
I therefore had estimates both of AF consumption and excretion and was able to show that AF metabolites only appeared in the urine samples of those individuals consuming the AF-contaminated peanut butter.
11
We also found that consumers of AF-contaminated foods were excreting AF metabolites in their urine that proved carcinogenic
12
to animal test subjects.
13
Throughout this research period, I continued to believe, as other researchers did, that AF might be an important carcinogen for humans. But I also understood that this very potent animal carcinogen had not yet been shown to be a human carcinogen—at least not in an independent manner. We knew at that time, for example, that the mouse, unlike the rat, was not susceptible to AF carcinogenicity,
14
and if these closely related species responded to AF in totally opposite ways, one susceptible and one resistant, it was not unreasonable to assume that humans might also be resistant as well. Clearly we still had a lot to learn about AF’s connection to cancer: was it relevant to humans, and if so, what was the causal mechanism?
15
In exploring these questions, I started with the assumption that the MFO enzyme was involved because evidence suggesting its relationship to AF and cancer already had been published by a research group in England.
16
It showed that MFO was responsible for converting AF into not one but several less carcinogenic products that were excreted in milk and urine. The more efficiently MFO functioned (i.e., the more “active” it was), the more AF was detoxified, suggesting that increasing MFO activity might lower the risk of liver cancer.
At around the same time, researchers were discovering that MFO’s activity could be modified—sped up, slowed down, and altered in other
ways—by certain agents, like drugs.
17
In my laboratory, we were finding that increasing dietary protein increased MFO activity.
18
Perhaps, we thought, protein could be used to supercharge MFO and stop cancer in its tracks.
Then I stumbled upon that 1968 report from India I mentioned in
chapter three
that showed what appeared to be the opposite: namely, that higher dietary protein
increased
AF-induced tumor development.
19
That couldn’t be! Protein, everyone’s favorite nutrient, could cause cancer? And the protein they used was casein, the principal protein in the healthiest drink there was: cow’s milk. I needed to learn more about this finding and either reproduce it or refute it as a fluke.
At the same time I was discovering an equally unsettling fact about childhood liver cancer in the Philippines: it occurred with much higher frequency not necessarily in the children who consumed greater quantities of AF, but in children from wealthier families, the ones who ate more protein and more “high quality” animal protein. The Indian protein/tumor study and the Philippine animal protein/cancer connection were starting to shake my world. Did more protein prevent cancer or cause cancer?
The possible key to solving this mystery was MFO, the startling enzyme that was now implicated both in the initiation of liver cancer by AF and in the detoxification and disposal of AF from the body. What was going on? Did dietary protein speed up MFO’s conversion of AF into nontoxic water-soluble metabolites? Or did it activate AF into nasty carcinogenic metabolites? Or both? We suspected we were on to something much bigger than just a way to neutralize or promote AF-induced liver cancer. We theorized that MFO might be a key factor in turning cancer on and off not just in the liver, but possibly also in other tissues in the human body.
This paradoxical protein effect hinted at what we eventually found to be the case: MFO responds to the foods that we eat every day. Certain diets turn MFO into a highly efficient cancer-fighting machine; other diets send MFO into a frenzy that produces carcinogenic by-products.
To understand how this is possible, we need to look at nutrition and how it affects enzymes more generally. Not only will we resolve the MFO–AF paradox, we’ll also see how reductionist nutritional thinking simply can’t handle the question—and thereby misses the most powerful lever we possess in our effort to eradicate cancer.
If you took high school biology, you probably spent some time memorizing bits of a chart of aerobic respiration known as the Krebs cycle. That chart, if it didn’t put you to sleep first, probably gave you the idea that nutrition is a very linear process. From the inputs of carbohydrates, fats, and proteins, the cells in the body predictably extract energy, produce a myriad collection of useful metabolites, and release leftover carbon dioxide and water. The arrows that connect different steps in the process seem authoritative, as if the described step always happens in precisely the same way every place, every time, under every condition. While this model is useful for understanding the basics, it doesn’t reliably correspond to reality. Nutrition is far more complex than a static diagram might imply.
Nutrients generally do not follow a single predictable path after they enter the trillions of cells in our bodies. In most cases, the potential route a nutrient can take once it enters the body branches out, directly or indirectly, into multiple pathways of products (metabolites), with each pathway possibly branching out into still more pathways. Furthermore as these pathways develop, they may lead to many different kinds of activities or functions, like mobilization of energy and repair of damaged cells. The dominant pathways end up determining to a great extent whether we enjoy health or suffer disease. Understanding metabolism is not just a matter of following a nutrient down a large number of independent pathways, however. As these pathways branch out, their integration with one another seems endless.
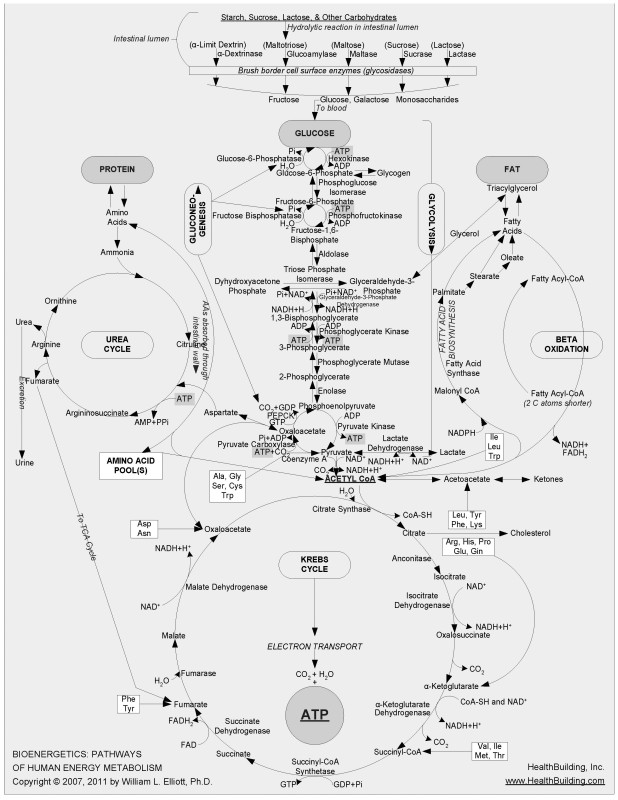
FIGURE 7-1.
Chart mapping glucose metabolism and other metabolic pathways
20
Maps of these metabolic mazes decorate the walls of many research facilities; your high school Krebs cycle chart is just a highly pared-down version of part of one of them. I’ve been in this research business long enough that I’ve been able to watch the emergence of one of the most complex of these maps, which began many years ago as the glucose-metabolism network of reactions that produces energy shown in
Figure 7-1
. (This particular chart, which does an excellent job of displaying the complexity of intermediary metabolism, is the work of Dr. William L. Elliott [
HealthBuilding.com
].) The earliest version of this map was most helpful as I taught biochemistry during the 1960s and 1970s at Virginia Tech’s Department of Biochemistry and Nutrition. It took me at least a dozen lectures in a basic biochemistry course merely to describe the series of
reactions that lead from glucose to the circular Krebs cycle at the bottom of the chart, primarily representing the extraction of energy from glucose.
Complicated, right? But the map I used in class only scratched the surface of what we know now about glucose’s metabolic pathway. Over time, more clusters of metabolic reactions were added to that initial map, including segments on protein, fat, and nucleic acid metabolism. It wasn’t long before so many reactions had been added, and the font size had become so small for reasonably sized paper, that it was clear that no more could be added and still be readable by the naked eye. The metabolic cartographers began creating entire atlases of cellular metabolism, with what had once been simple reactions now meriting several pages of diagramming to account for updated discoveries.
These comprehensive maps became more and more specialized and fragmented in a way that graphically symbolizes how reductionism, by pushing for ever smaller and more specific pieces of information, loses sight of the whole. Researchers spent years, even decades, working on just one or two reactions. Gradually, insets of insets of insets emerged on the map, as our probes of knowledge went ever deeper into cellular metabolism and grew ever less able to see the intelligence and power of the whole system.
A phrase with the same root as
reductionism
is “reductio ad absur-dum,” or following a concept to the point of absurdity. Remember
Figure 7-1’s
complex chart showing glucose metabolism? You can see an updated version in
Figure 7-2
.
Scientists have gone even deeper than this.
Figure 7-3
shows the complexity involved in just a very small section of that map, blown up for visibility.
And the more comprehensive metabolic map in
Figure 7-2
is only an infinitesimally small portion of all the reactions in each of our hundred trillion cells.
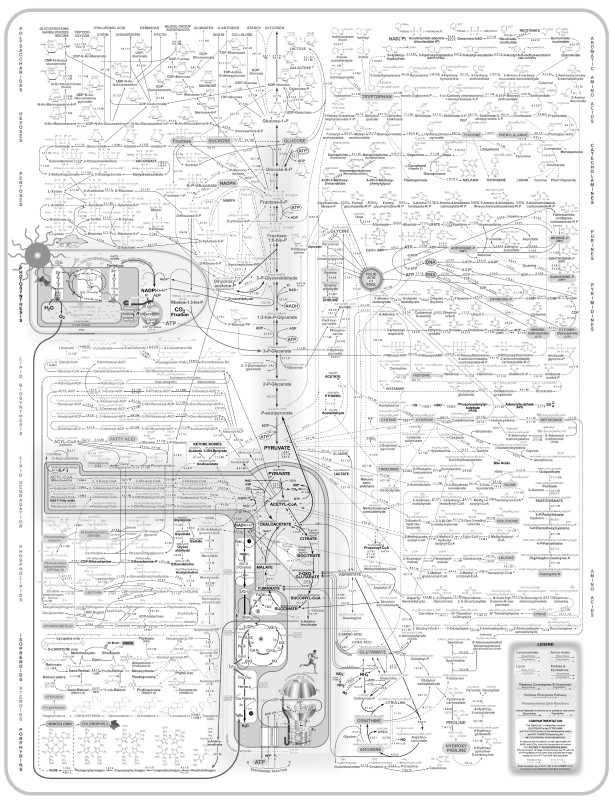
FIGURE 7-2.
Expanded chart mapping glucose metabolism and other metabolic pathways