Examination Medicine: A Guide to Physician Training (45 page)
Read Examination Medicine: A Guide to Physician Training Online
Authors: Nicholas J. Talley,Simon O’connor
Tags: #Medical, #Internal Medicine, #Diagnosis

This is a disseminated malignant disease of plasma cells. It can present as a diagnostic or a management problem. Myeloma occurs more commonly in the elderly – the median age is 60 years – and more often in men. It is more common in people whose occupations involve exposure to petroleum and in people exposed to nuclear radiation. Chronic antigenic stimulation may play a role in B cell clonal transformation. A number of chromosomal deletions and translocations have been identified in myeloma patients. The most important of these is 13q; its presence has prognostic significance. Some myeloma begin as monoclonal gammopathies of uncertain significance (MGUS). MGUS are much more common than myeloma – they are found in up to 10% of 80-year-olds – and they evolve to myeloma at a rate of 1% a year (‘smouldering myeloma’).
The history
1.
Ask about presenting symptoms:
a.
bone pain or pain with movement affects nearly three-quarters of patients, occurs particularly in the ribs or axial skeleton and may cause pathological fractures; the association with movement may distinguish it from the bone pain of metastatic malignancy, which can be worse at night (persistent pain suggests a pathological fracture)
b.
bacterial infection is the presenting problem in one-quarter of patients particularly pneumonia and urinary tract infections (total immunoglobulins are increased, but the level of normal functional immunoglobulins is reduced as a result of reduced production and increased destruction; in advanced marrow disease there is white cell depletion); the CD4 count is low; the most common organisms are
Streptococcus pneumoniae
,
Staphylococcus aureus
and
Klebsiella pneumoniae
in the lungs, and
E. coli
in the urinary tract
c.
symptoms of anaemia (normochromic and normocytic) – as a result of bone marrow depression from infiltration, chronic disease, renal failure or treatment
d.
bleeding tendency – caused by paraprotein inactivation of plasma procoagulants and reduced platelet function (coating of platelets with antibodies) and thrombocytopenia as a result of bone marrow suppression
e.
renal disease symptoms (owing to stone formation secondary to hypercalcaemia, hyperuricaemia, tubular damage by light chains, therapy, urinary tract infection, contrast studies, plasma cell infiltration or amyloid) – renal failure affects one-quarter of patients and half develop renal impairment; renal impairment at the time of diagnosis means a poor prognosis
f.
hypercalcaemic symptoms owing to bone lysis
g.
hyperviscosity symptoms once plasma viscosity exceeds 5 (normal is about 1.8)
h.
spinal cord compression or, rarely, diffuse sensorimotor neuropathy
i.
skin changes, such as pruritus, purpura, yellow skin, hypertrichosis (rare), erythema annulare (rare)
j.
systemic amyloid deposition (10–15%).
2.
Find out how the diagnosis was made. This nearly always requires biopsy of the marrow or an extramedullary plasmacytoma.
3.
Ask about treatment – ask about drugs (e.g. bisphosphonates, thalidomide) and their side-effects, radiotherapy and bone marrow transplant (actual or planned).
4.
Enquire about the patient’s social history, including dependants, work, ADLs etc.
5.
Determine the patient’s understanding of this life-threatening condition and its prognosis – how much information he or she has about possible further problems and treatment.
The examination
1.
Inspect the patient for signs of weight loss, anaemia, chest infection and general debility.
2.
Examine the haemopoietic system. Pay particular attention to a search for bony tenderness. Kyphosis may be caused by compression fractures. Splenomegaly and lymphadenopathy occur only very rarely.
3.
Note signs of anaemia and purpura. Look for skin rash.
4.
Look for signs of infection (e.g. pulmonary consolidation).
5.
Check carefully for any signs of spinal cord compression.
6.
Check the urine analysis and temperature chart.
Investigations
1.
Once the diagnosis is suspected, check the full blood count (and film) for anaemia (and rouleaux) and a raised ESR. Obtain a protein electrophoretogram (EPG) of serum and urine and an immunoelectrophoretogram (IEPG). An ‘M’ component (monoclonal globulin peak) is found in 95% of cases (any immunoglobulin class may appear as the ‘M’ component). Light chains are present in the urine in 50–75% of patients (Bence-Jones proteinuria cannot be detected by dipstick urine analysis).
2.
Analysis of circulating light chains in the serum is a more sensitive way of detecting light chain myeloma as an abnormal κ:λ ratio is seen. It is helpful in monitoring the response to treatment of such patients.
3.
The bone marrow must be examined for plasma cells (>10% in myeloma). Check the beta2-microglobulin level (see below).
4.
Look at X-ray films of the skull (
Fig 8.4
), chest, pelvis (
Fig 8.5
) and proximal long bones for fractures, osteoporosis and lytic lesions. The latter is a result of the secretion of osteoclast-activating factors by the tumour cells. Since there is little osteoblastic activity, bone scans are much less sensitive than plain X-rays.
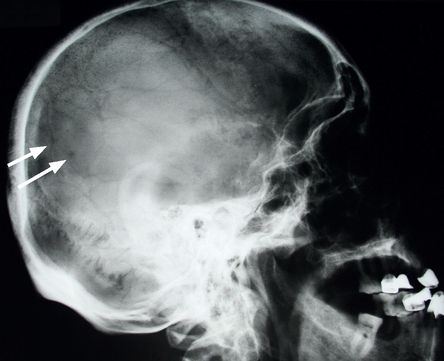
FIGURE 8.4
Skull X-ray of a myeloma patient showing multiple lucent areas pepper-pot skull (arrows). Figure reproduced courtesy of The Canberra Hospital.
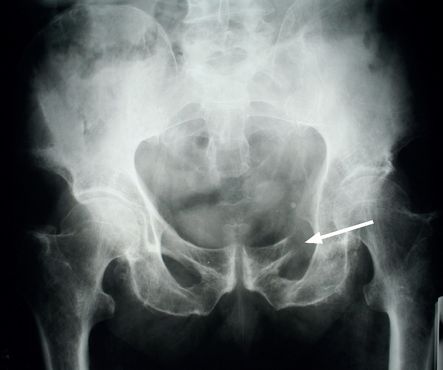
FIGURE 8.5
X-ray of the pelvis showing destruction of the left superior ramus of the ischium (arrow). Figure reproduced courtesy of The Canberra Hospital.
5.
MRI scans may be the best way to investigate pain and possible nerve root or spinal cord compression.
6.
Also check the serum calcium and urate levels and the renal function.
The three major diagnostic features of multiple myeloma, in order of importance, are:
1.
plasma cells in the bone marrow (>10% involvement is consistent with the diagnosis)
In the marrow there will be >10% plasma cells with evidence of at least organ/tissue injury: CRAB (Hyper Calcaemia, Renal disease, Anaemia and Bone lytic lesions)
2.
production of serum paraprotein (50% are IgG, 33% IgA, 5% IgM, 10% only light chains, 2% nil)
3.
bone destruction (lytic lesions).
The disease can be staged according to certain criteria (
Table 8.20
). On this basis, median survival can be estimated as follows:
Table 8.20
International staging system
| 1. Stage 1 | Beta 2 microglobulin < 3.5mg/dL and serum albumin ≥ 3.5g/dL |
| 2. Stage II | Neither I or II |
| 3. Stage III | Beta 2 microglobulin ≥ 5.5mg/L |
The International Myeloma Foundation.
•
stage I – 62 months (median)
•
stage II – 44 months (median)
•
stage III – 29 months (median).
An elevated serum beta2-microglobulin level (which reflects the myeloma cell burden and renal functional impairment) indicates a reduced median survival. Other poor prognostic features include advanced age and cytogenetic abnormalities (e.g. any translocation, deletion of chromosome 13q).
Treatment
1.
Irradiation is helpful for localised bone pain and spinal cord compression. Patients with a single bone plasmacytoma will often get prolonged disease-free survival after treatment with local radiotherapy. Pamidronate or one of the other bisphosphonates should be given to patients with more than stage I disease. Bisphosphonates reduce bone pain, fracture rates and episodes of hypercalcaemia. There is evidence that they improve the prognosis.
2.
For those with more diffuse disease, general measures, such as adequate hydration and use of bicarbonate for Bence-Jones proteinuria, are important to prevent renal failure. Intravenous contrast material must be used cautiously and only with excellent hydration. Allopurinol may protect renal function from urate nephropathy related to treatment. Treat hypercalcaemia and bacterial infection. Avoid live vaccines. Erythropoietin may improve the anaemia.
3.
Systemic therapy is indicated for patients with stage II or III disease and for those with stage I disease if they are symptomatic or have rising myeloma protein levels or progressive lytic bone lesions. Treatment is begun with high-dose steroids in combination with thalidomide or lenalidomide (reduce tumour necrosis factor alpha). An alternative is bortezomib, a proteosome inhibitor.
4.
Alkylating agents should be avoided if the patient may be a candidate for bone marrow transplant. Such drugs may prevent stem cell harvesting by damaging these cells. Melphalan and prednisone were standard treatment; other alkylating agents (e.g. cyclophosphamide) are probably equally effective for patients unsuitable for bone marrow transplant. Resistance to one alkylating agent is often, but not always, associated with resistance to the others.
Older patients (ineligible for autologous transplant) typically receive melphalan, thalidomide and prednisone (MTP) orally. Bortezomib, melphalan and prednisone (VMP) is an alternative. Typically treatment is given intermittently. A week’s course is repeated 4 weeks later for up to 2 years. The patient’s progress must be monitored with regular protein EPG studies.
5.
Relapse is treated for suitable patients with autologous stem cell bone marrow transplant.
DIFFERENTIAL DIAGNOSIS
Monoclonal gammopathy of undetermined significance
A smaller monoclonal peak (<30 g/L), fewer than 10% plasma cells in the bone marrow and absence of lytic bone lesions suggest this diagnosis. Urinary light chains are absent. Infection, renal failure and anaemia are not increased. No therapy is required.
Waldenström’s macroglobulinaemia
The EPG has a peak consisting of monoclonal IgM. These patients are generally older than those with myeloma. The hyperviscosity syndrome is often present; symptoms and signs include lassitude, confusion, bleeding, anaemia, infection, lymphadenopathy and splenomegaly, dilated retinal veins and perivenous haemorrhages, and (rarely) renal failure. Lymphadenopathy and splenomegaly do not usually occur in patients with myeloma. Lytic bone destruction and hypercalcaemia are rare. Ten per cent of the macroglobulins are cryoglobulins. An underlying lymphoproliferative disorder may be present. Treatment with plasmapheresis is effective in removing IgM paraprotein. Prednisone, fludarabine and chlorambucil are useful. Rituximab (anti-CD20 monoclonal antibody) and thalidomide have also been used successfully. Median survival is 4 years.
Localised myeloma
Only one plasma cell tumour is present. Solitary plasmacytomas often occur in the nasopharynx or paranasal sinuses. The major complications of myeloma are absent. Only 50% of cases show a monoclonal peak. Local radiotherapy is the usual treatment.
POEMS syndrome
This is an atypical form of myeloma. The features include:
P
olyneuropathy
O
rganomegaly
E
ndocrinopathy
M
onoclonal gammopathy (osteosclerotic myeloma, or IgA or IgG M proteins with lambda light chains)
S
kin changes
There is a progressive sensorimotor polyneuropathy associated with myeloma-like bone lesions. Unlike multiple myeloma patients, over half these patients have hepatomegaly and lymphadenopathy and some have splenomegaly. Male erectile dysfunction and gynaecomastia and female amenorrhoea occur as a result of hyperprolactinaemia. A few patients have hypothyroidism and one-third have type 2 diabetes. Skin changes include clubbing, hypertrichosis, thickening and increased pigmentation.