Pediatric Examination and Board Review (156 page)
Read Pediatric Examination and Board Review Online
Authors: Robert Daum,Jason Canel

(C) coronary artery disease
(D) restrictive lung disease
(E) growth retardation
16.
Which of the following types of chemotherapy is not matched correctly with one of its side effects?
(A) doxorubicin and pulmonary fibrosis
(B) cisplatin and high-frequency hearing loss
(C) cyclophosphamide and hemorrhagic cystitis
(D) corticosteroids and avascular necrosis
(E) vincristine and neuropathy
ANSWERS
1.
(D)
Osteosarcoma and Ewing sarcoma are both primary bone malignancies that occur in children and young adults and most commonly present with pain localized to the tumor site. Osteosarcoma (also known as osteogenic sarcoma) is a relatively common pediatric malignancy, with approximately 400 cases per year in children younger than 20 years of age in the United States. Ewing sarcoma and a related tumor, peripheral primitive neuroectodermal tumor (PPNET), are similar entities with similar tissues of origin that can arise within either bone or soft tissues and are members of a group of tumors called the “small round blue cell tumors of childhood,” a group that also includes neuroblastoma, lymphoma, and rhabdomyosarcoma. Ewing sarcoma is one of the few solid tumors for which the underlying molecular genetic abnormality has been described: rearrangement of the
EWS
gene on chromosome 22q12 with an
ETS
gene family member. In 95% of cases, a t(11;22)(q24;q12) translocation is detected. These translocations define the Ewing sarcoma family of tumors (ESFT) and provide a valuable tool for their accurate and unequivocal diagnosis. They also represent ideal targets for the development of tumor-specific therapeutics. Ewing sarcoma accounts for approximately 100 cases of pediatric malignancy per year in the United States. Osteoid osteoma is a common benign bony lesion that generally occurs in the lower extremities and presents with night-time pain that is relieved with nonsteroidal anti-inflammatory medications. Nonossifying fibromas (also called fibrous cortical defects) are benign developmental defects in ossification that are painless, usually only detected incidentally, and require no therapy (see
Figure 90-1
).
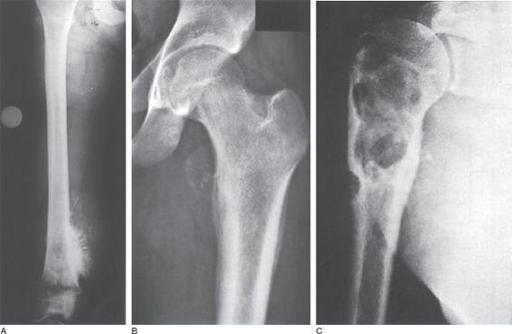
FIGURE 90-1.
X-ray imaging of osteosarcoma, Ewing sarcoma, and chondrosarcoma.
A.
The typical “sunburst” appearance of osteosarcoma.
B.
The “onionskin” appearance often seen in Ewing sarcoma.
C.
The lobulated appearance of chondrosarcoma. (Reproduced, with permission, from Kantarjian HM, Wolff RA, Koller CA. MD Anderson Manual of Medical Oncology. New York: McGraw-Hill; 2006: Fig 33-7A-C.)
2.
(A)
Osteosarcoma most commonly occurs in the metaphyses of long bones but can spread into the diaphyses and epiphyses via local invasion. Bone marrow involvement with osteosarcoma is extremely rare.
The most common sites of osteosarcoma are at those sites with the most rapid bone growth: the distal femur, proximal tibia, and proximal humerus. Approximately 65% of all osteosarcomas occur in the femur, and more than 80% of femoral osteosarcomas occur in the distal end. The second most common site is the proximal tibia; the third most common site is the proximal humerus. Approximately 20% of osteosarcomas occur in the arms. In general, the outcomes for localized osteosarcoma are good, with 60-80% long-term survival with current treatments. Children with metastatic osteosarcoma, however, do significantly worse, with only a 20-30% survival rate. Osteosarcoma can also rarely occur in flat bones such as the skull, ribs, and pelvis, where it also has a much poorer outcome.
3.
(D)
The peak incidence of osteosarcoma occurs in the second decade of life, particularly during the pubertal growth spurt, and tends to occur slightly earlier in girls (who have an earlier onset of puberty). The development of osteosarcoma may in part be associated with the rapid bone growth that occurs during these times. Osteosarcoma is very rare in children younger than 5 years of age and uncommon in adults older than 30 years of age.
4.
(C)
Down syndrome is associated with an increased incidence of hematologic malignancies but does not have an increased incidence of osteosarcoma or other sarcomas. Children with cases of bilateral retinoblastoma almost always have germ-line mutations in the
Rb
gene, which are also associated with an increased incidence of secondary malignancies, approximately 50% of which are osteosarcomas. The risk of osteosarcoma in patients with
Rb
gene mutations is approximately 500 times that of the general population.
Prior radiation exposure, including radiation therapy for childhood malignancies, is also associated with an increased incidence of osteosarcoma. Radiation exposure has been linked to up to 5% of osteosarcoma cases, and osteosarcoma can occur up to 40 years after exposure to radiation.
A family history of sarcomas, leukemias, adrenocortical carcinomas, and breast and bone cancers can be found in families with hereditary mutations in the
p53
gene, termed the Li-Fraumeni syndrome, which also is associated with an increased incidence of osteosarcoma. Other syndromes associated with increased osteosarcoma incidence include Paget disease and Ollier disease (enchondromatosis), both of which more commonly are associated with adult-onset osteosarcoma.
5.
(C)
Ewing sarcoma is most common during the second decade of life but can occur in younger and older populations as well. Approximately 70% of the cases occur in children younger than 20 years of age, and half of the cases occur between 10 and 20 years of age. Ewing sarcoma is extremely rare in adults older than 30 years of age. Ewing sarcomas are also extremely rare in African Americans and Asians, occurring most commonly in white populations. There is no apparent connection between the onset of Ewing sarcoma and the occurrence of puberty, and there are no associated syndromes or exposures that increase the risk of Ewing sarcoma.
6.
(D)
Ewing sarcoma can present in a wide variety of locations, and, although most arise from within the skeleton, some Ewing sarcomas can arise in soft tissues as well. The primary sites for Ewing sarcoma are the pelvis and lower extremities, with approximately 20% of cases in the pelvis, 20% in the femurs, and 10% each in the tibias and fibulas. Approximately 9% of cases occur in the chest wall and are sometimes known as Askin tumors. Ewing sarcomas are more common in the axial skeleton than osteosarcomas, but approximately 3% of cases occur in the skull.
7.
(D)
Ewing sarcoma and osteosarcoma both occur predominantly in the same age range of patients (between 10 and 20 years of age). However, Ewing sarcoma and osteosarcoma have distinct features that can assist in the diagnosis before biopsy. Ewing sarcoma tends to be associated with systemic symptoms such as fever and weight loss, whereas osteosarcoma usually presents with local symptoms such as pain and swelling. Ewing tumors are more commonly located in the axial skeleton and are usually diaphyseal, as opposed to osteosarcomas that are more commonly metaphyseal and more likely to occur in the extremities. Furthermore, Ewing sarcoma classically has associated reactive bone formation (described as “onion skin” or “hair-on-end” periosteal reaction visible on x-rays) that is not usually found in osteosarcomas, which typically present with lytic bony lesions. A family history of other sarcomas, leukemias, breast cancers, or adrenal cancers suggests the possibility of Li-Fraumeni syndrome, a tumor predisposition syndrome which occurs as a result of mutations in the
p53
gene. It is associated with an increase in the incidence of osteosarcoma but is not associated with Ewing sarcoma.
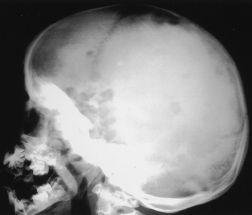
FIGURE 90-2.
Extensive Langerhans cell histiocytosis. Radiograph of the skull shows well-circumscribed osteolytic areas with a typical “map” appearance. (Reproduced, with permission, from Wolff K, Goldsmith LA, Katz SI, et al. Fitzpatrick’s Dermatology in General Medicine, 7th ed. New York: McGraw-Hill; 2008: Fig. 148-11.)
8.
(A)
Langerhans cell histiocytosis (LCH) is a monoclonal disorder of histiocytes with a wide variety of clinical presentations that affects approximately 4 children per million per year (see
Figure 90-2
). Previously recognized entities such as histiocytosis X, Letterer-Siwe disease, Hand-Schüller-Christian syndrome, and eosinophilic granuloma are now all classified as subtypes of LCH. The most benign form of LCH is eosinophilic granuloma, which consists of isolated lytic bony lesions, most commonly in the skull but also occurring in the vertebrae, mandible, ribs, ilium, scapula, and long bones. The bony lesions can be asymptomatic or can cause localized pain and swelling. Hand-Schüller-Christian disease is multifocal LCH characterized by skull lesions, diabetes insipidus, and exophthalmos. Other less common features include other pituitary hormonal abnormalities, gingival ulcerations with premature tooth eruption, chronic otitis media, and persistent seborrheic rashes. Letterer-Siwe disease is the most severe form of LCH and usually has an onset before 2 years of age. Letterer-Siwe disease is characterized by more severe visceral involvement, including lung, liver, intestinal, and marrow disease, with persistent fevers, irritability, failure to thrive, malabsorption, pancytopenia, and other symptoms related to diffuse organ involvement. Letterer-Siwe disease only accounts for 15% of LCH cases but has the worst prognosis and is the least responsive to therapy.
9.
(B)
Hemophagocytic lymphohistiocytosis (HLH) is a histiocytic syndrome that can be either primary or secondary to infection or neoplasia. Proliferation of activated macrophages results in the symptoms of HLH, which can involve the liver, spleen, bone marrow, and central nervous system. Symptoms include fever, hepatosplenomegaly, skin rashes, and meningeal inflammation with meningismus and, potentially, seizures. Laboratory features include pancytopenia, hypertriglyceridemia, hypofibrinogenemia, hyperferritinemia, and hypoproteinemia. Depressed T-cell and NK-cell activity can also be demonstrated. Alkaline phosphatase levels are generally normal. Diagnosis of HLH requires the presence of fever, splenomegaly, peripheral cytopenias of at least 2 cell lines, hypertriglyceridemia or hypofibrinogenemia, and evidence of hemophagocytosis either in the bone marrow or in a lymph node biopsy specimen.
10.
(C)
Blood products for patients receiving chemotherapy should be leuko-reduced and irradiated to decrease the transmission rate of cytomegalovirus (carried by donor white blood cells) and to decrease the incidence of graft-versushost disease (mediated by viable donor lymphocytes). Exposure of the patient to blood products from relatives should be avoided to prevent alloimmunization of the patient to potential bone marrow donor antigens. Furthermore, patients receiving chemotherapy often require frequent blood product transfusions, and so exposure to unrelated donors should be minimized as much as possible. Therefore, platelet apheresis units, which are isolated from single donors, are preferred over units pooled from multiple donors. FFP should be given to replace coagulation factors when needed and should be blood-type matched to reduce the incidence of immune-mediated reactions from the antibodies carried in the transfused plasma.
11.
(A)
Platelet transfusions are necessary both for control of active bleeding and for prevention of spontaneous hemorrhages in patients with decreased or dysfunctional platelets. Platelet transfusions should be given to prevent spontaneous hemorrhages when the platelet count is lower than 10,000/μL. In most cases, a lumbar puncture can be performed safely with platelet counts higher than 10,000/μL. Intramuscular injections should be avoided when platelet counts are less than 20,000/μL. To prevent intracranial hemorrhages in patients with brain tumors, the platelet count should be higher than 30,000-50,000/μL.